|
Thực hành tốt thử nghiệm lâm sàng và vấn đề về đạo đức trong các nghiên cứu y sinh học
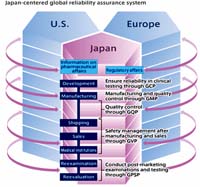
Thử nghiệm lâm sàng là một phần không thể thiếu hiện nay trong bối cảnh phát triển khoa học và công nghệ trên tất cả các ngành khoa học, đặc biệt là lĩnh vực ydươc học,đặc biệt hơn khi chúng ta áp dụng nghiên cứu trên chính cơ thể con người. Chính vì lẽ đó các nghiên cứu cần phải tuân thủ các tiêu chuẩn nghiêm ngặt, đặc biệt theo quy định của các tổ chức quốc tế và nước sở tại và đặc thù của từng loại thiết kế nghiên cứu để làm sao vừa dạt được mục nghiên cứu, kết quả với những số liệu đáng tin cậy nhưng không được làm tổn hại đến người tham gia nghiên cứu . Mối liên hệ giữa thầy thuốc-bệnh nhân trong các nghiên cứu lâm sàng là một lĩnh vực y đức, nhưng chưa được bàn thảo rộng rãi ở nước ta. Những tiến bộ ngoạn mục của y học hiện đại, kể cả thuốc mới, không thể có được nếu không có sự tham gia tự nguyện của bệnh nhân trong các nghiên cứu lâm sàng. Trong các nghiên cứu này, bệnh nhân thường được tuyển chọn theo những tiêu chuẩn chọn đàu vào và họ được theo dõi một thời gian để đánh giá hiệu quả và an toàn của một thuật điều trị. Trong quá trình theo dõi, bệnh nhân phải tốn nhiều thì giờ đến tái khám, hay trong nhiều trường hợp phải cung cấp các mẫu máu, nước tiểu hoặc một mô nào đó trong cơ thể để bác sĩ làm xét nghiệm. Nói cách khác, bệnh nhân tham gia vào những nghiên cứu lâm sàng thường phải hi sinh thời gian và có khi thủ thuật nghiên cứu mang tính xâm phạm. Do đó, mục tiêu và phương pháp nghiên cứu phải được xem xét cẩn thận sao cho quyền lợi và lợi ích của bệnh nhân nhận ưu tiên số một theo đúng tinh thần của nguyên tắc y khoa là "trước hết, không làm hại" (primum non nocere). Trong phạm vi bài viết, xin trình bày nội dung thực hành thử nghiệm lâm sàng tốt và khía cạnh đạo đức trong các nghiên cứu y sinh học, đặc biệt trên người. Bài viết này có tham khảo tài liệu trong và ngoài nước, đặc biệt bài biết của PGS.TS. Nguyễn Thị Kim Tiến, Chủ tịch Hội đồng Khoa học, Chủ tịch Hội đồng đạo đức, Thứ trưởng Bộ Y tế. Mở đầu Trong xu thế hội nhập với thế giới và sự phát triển như vũ bão hiện nay của khoa học công nghệ nói chung và của ngành y - dược nói riêng, tại Việt Nam ngày càng có nhiều các sản phẩm thuốc mới (bao gồm thuốc tân dược, vaccin, thuốc y học cổ truyền, chế phẩm sinh học sử dụng cho điều trị) được đưa vào nghiên cứu thử nghiệm lâm sàng (thử nghiệm trên con người) do các nhà sản xuất, các hãng bào chế nước ngoài và trong nước đề nghị thử nghiệm. Bộ Y tế mà đầu mối là Vụ Khoa học và Đào tạo đã và đang là cầu nối giữa nhà quản lý, nhà sản xuất và các nhà nghiên cứu đối với nghiên cứu thử nghiệm lâm sàng với mục tiêu tạo hành lang pháp lý, ban hành các tiêu chuẩn kỹ thuật cho việc nghiên cứu, phát triển, ứng dụng những sản phẩm mới bảo đảm an toàn, hiệu quả, bảo vệ quyền lợi và sức khoẻ cho người tham gia vào nghiên cứu, tạo điều kiện cho người dân được tiếp cận với những thành quả của sự phát triển khoa học kỹ thuật nói chung và những sản phẩm là thuốc nói riêng. Liên quan đến việc thực hành tốt thử nghiệm lâm sàng và các vấn đề đạo đức trong nghiên cứu, bài viết này gồm các nội dung chủ yếu sau: khái niệm thử nghiệm lâm sàng thuốc, các nội dung đạo đức trong nghiên cứu y sinh học và Hội đồng đạo đức trong nghiên cứu y sinh học, ngành y tế Việt Nam hội nhập với quốc tế trong lĩnh vực nghiên cứu lâm sàng và các quy trình, thủ tục chủ yếu trong quá trình thực hiện nghiên cứu thử nghiệm lâm sàng tại Việt Nam. Thử nghiệm lâm sàng thuốc là gì? Thử nghiệm lâm sàng (TNLS) thuốc là hoạt động khoa học nghiên cứu một cách hệ thống trên người nhằm đánh giá hiệu quả lâm sàng, nhận biết và phát hiện các phản ứng bất lợi, nghiên cứu sự hấp thu, phân bố, chuyển hoá và sự thải trừ của thuốc nhằm mục đích chứng minh sự an toàn và hiệu quả của thuốc thử nghiệm. Thuật ngữ thử nghiệm lâm sàng thuốc hay nghiên cứu lâm sàng (NCLS) và / hoặc thử thuốc trên lâm sàng (TTTLS) là đồng nghĩa với nhau. Thử nghiệm lâm sàng thuốc là quá trình nghiên cứu phức tạp đòi hỏi sự đầu tư thích đáng, lâu dài về thời gian, công sức và kinh phí. Trung bình đối với một nghiên cứu thuốc mới, thời gian nghiên cứu lâm sàng kéo dài từ 5 - 10 năm và trải qua 4 giai đoạn (phase): Giai đoạn 1: -Là giai đoạn lần đầu tiên thử sản phẩm thuốc mới trên người; -Mục đích giai đoạn này là thiết lập sự đánh giá sơ bộ về tính an toàn, dược lực học và dược động học của thuốc trên đối tượng là con người; -Giai đoạn 1 có thể có một số bằng chứng sớm về tính hiệu quả; -Tổng số đối tượng ở giai đoạn 1 có thể từ 20 đến 80 đối tượng. Giai đoạn 2: -Là giai đoạn thử nghiệm được tiến hành trên số lượng đối tượng hạn chế, nhưng với số lượng lớn hơn giai đoạn 1 có thể từ một đến vài trăm đối tượng. -Mục đích nghiên cứu giai đoạn 2 nhằm đánh giá tác dụng trị liệu, tính an toàn của thuốc trên các bệnh nhân có bệnh, xác định liều lượng, cách dùng thích hợp để đưa ra trị liệu tối ưu cho TNLS. -Nghiên cứu thiết kế so sánh ngẫu nhiên, có đối chứng. Giai đoạn 3: -Là giai đoạn TNLS được tiến hành trên nhóm đối tượng lớn hơn giai đoạn 2, số lượng đối tượng có thể từ vài trăm đến vài ngàn đối tượng; -Mục đích nghiên cứu giai đoạn 3 nhằm xác định độ ổn định của thuốc, độ ổn định của công thức thuốc, tính an toàn, hiệu quả ngắn hạn và dài hạn của thuốc, đánh giá giá trị trị liệu ở mức tổng thể, nghiên cứu các phản ứng bất lợi thường xuyên xảy ra, phát hiện các đặc điểm đặc biệt của sản phẩm nghiên cứu; -Nghiên cứu được thiết kế ngẫu nhiên đơn hoặc kép; -Các điều kiện TNLS trong giai đoạn này được tiến hành gần với điều kiện sử dụng thực tế; -TNLS giai đoạn 3 cung cấp cơ sở khoa học cuối cùng để có thể giấy phép sản xuất đưa ra thị trường nếu kết quả TNLS là khả quan. Giai đoạn 4: -Giai đoạn 4 là các nghiên cứu TNLS được tiến hành sau khi thuốc đã được đưa vào lưu hành; -Giai đoạn 4 còn được áp dụng để thiết kế các TNLS nhằm đưa ra các chỉ dẫn mới về cách dùng thuốc, phương pháp dùng mới hoặc kết hợp mới, tương tự như những TNLS cho sản phẩm thuốc mới; Tuy nhiên đối với các trường hợp TNLS nêu trên không bắt buộc phải quay lại từ giai đoạn 1 mà nó kế thừa các kết quả nghiên cứu ở các giai đoạn trên (nghiên cứu "bắc cầu"). Có thể tóm tắt quá trình nghiên cứu một thuốc mới theo một sơ đồ đơn giản Thực hành tốt thử nghiệm lâm sàng là gì? Thực hành tốt thử nghiệm lâm sàng còn được dịch là "Thực hành lâm sàng tốt" (Good Clinical Practice - GCP) là một tài liệu hướng dẫn mang tính chuẩn mực quốc tế nhằm hướng dẫn cho nhà nghiên cứu, nhà tài trợ, các cơ quan quản lý, các hội đồng xét duyệt về đạo đức và khoa học trong thiết kế, tiến hành, thực hiện, theo dõi, giám sát, kiểm định, ghi chép, phân tích và báo cáo đối với các TNLS. Nó bảo đảm độ tin cậy, tính chính xác của các dữ liệu được báo cáo và sự chấp nhận mang tính quốc tế đối với các kết quả TNLS, đồng thời bảo đảm sự an toàn, quyền của các đối tượng nghiên cứu trong các TNLS. Các hướng dẫn quốc gia về các TNLS có giá trị áp dụng tại nước sở tại, nhưng chỉ được quốc tế công nhận các kết quả TNLS khi các hướng dẫn TNLS tại quốc gia đó tuân thủ theo chuẩn mực của GCP quốc tế ban hành như GCP của ICH (International conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human use - Hội nghị quốc tế về hài hoà sử dụng dược phẩm trên con người) hoặc GCP của WHO (World Health Organization - Tổ chức Y tế Thế giới). Hiện nay, các quốc gia trong khu vực đã ban hành hướng dẫn GCP của mình bao gồm: Singapore (năm 1998), Malaysia (năm 1999), Trung Quốc (năm 1999), Thái Lan (năm 2000), Indonesia (năm 2001), Việt Nam (năm 2008). Hướng dẫn Tổ chức Y tế thế giới về thực hành thử nghiệm lâm sàng tốt Tổ chức Y tế thế giới gần đây đã tổ chức một cuộc họp không chính thức như một bước đầu tiên trong tổng hợp hướng dẫn về thực hành thử nghiệm lâm sàng tốt trong cuốn WHO Guidelines on Good Clinical Practice (GCP) đối với các thử nghiệm trên các sản phẩm dược học. Các đại biểu tham gia đến từ Argentina, cộng đồng châu Âu, Trung Quốc, Indonesia, Nhật Bản, Jordan, Thụy Sĩ và Mỹ cùng nhau thống nhất các nội dung chương trình liên quan đến thực hành thử nghiệm lâm sàng tốt. GCP bao gồm các vấn đề thiết kế, thực hiện, giám sát, thanh tra, phân tích, báo cáo và trình bày các nghiên cứu thử nghiệm lâm sàng. Việc áp dụng tiêu chuẩn GCP cho các nghiên cứu sẽ đảm bảo nghiên cứu mang tính khoa học và thực hiện tốt vấn đề đạo đức trong nghiên cứu và các khía cạnh trên lâm sàng như chất lượng, độ an toàn và hiệu quả của sản phẩm dưới sự giám sát trình bày hợp lý. Với sự hỗ trợ của tổ chức nghiên cứu bệnh nhiệt đới (WHO/TDR) phối hợp nghiên cứu thử nghiệm lâm sàng trên các bệnh bị lãng quên (neglected diseases), nâng cao sức khỏe tại các vùng và các quốc gia có bệnh lưu hành. Để đảm bảo rằng các thử nghiệm lâm sàng đặc biệt trên người theo các tiêu chuẩn GCP quốc tế cao nhất, WHO-TDR đã thiết kế và tiến hành một số khóa học về GCP liên quan đến điều tra và giám sát về mặt thử nghiệm lâm sàng tại các quốc gia đang phát triển hơn 6 năm qua, để thúc đẩy và bổ sung đầy đủ các nguyên tắc của GCP để bảo vệ giá trị, quyền lợi, an toàn cho các đối tượng một cách tốt nhất cũng như bảo đảm về số liệu một cách đáng tin cậy. Các khóa học như thế diễn ra hàng năm và có cả khóa đào tạo lại (GCP refresher courses). Tại châu Á, các đại học -Viện Sức khỏe quốc gia Manila, Philippines phối hợp với tổ chức WHO-TDR, SIDCER/FERCAP để tổ chức một số hội thảo đào tạo về khía cạnh y đức trong nghiên cứu sức khỏe và thực hành thử nghiệm lâm sàng tốt nhằm góp phần nâng cao khả năng nghiên cứu chuẩn mực trong vùng. Hướng dẫn GCP (Good Clinical Practice Guidelines) bao gồm các tiêu chuẩn thế nào tiến hành một thửnghiệm lâm sàng, xác định vai trò và trách nhiệm của các thành viên cũng như người bảo trợ cho nghiên cứu lâm sàng, các nhà nghiên cứu và giám sát trong thử nghiệm lâm sàng. Trong đó vai trò giám sát khía cạnh công nghiệp dược gọi là Clinical Research Associates. Trong đó gồm đầy đủ các hướng dẫn liên quan đến người nghiên cứu (Guidelines for the investigator); hướng dẫn cho đề cương thử nghiệm lâm sàng và các phần phụ lục sửa đổi bổ sung cho đề cương (Guidelines for the clinical trial protocol and protocol amendments); hướng dẫn cho người tài trợ thử nghiệm lâm sàng (Guidelines for the trial sponsor) và hướng dẫn sử dụng cuốn sách thông tin cho người nghiên cứu (Guidelines for the Investigator's brochure) Quy trình cho một thử nghiệm lâm sàngthường liên quan đến tìm kiếm bệnh nhân đủ tiêu chuẩn, viết giấy cam kết tham gia nghiên cứu, nêu rõ mục đích nghiên cứu,…theo sơ đồ dưới đây: GCP_các sản phẩm thuốc trên người (Human Medicinal Products)GCP là một tiêu chuẩn và khía cạnh y đức liên quan đến thiết kế, ghi chép và báo cáo thử nghiệm lâm sàng liên quan đến đối tượng là con người tham gia vào nghiên cứu. Sự chấp thuận tham gia nghiên cứu chuẩn này sẽ đảm bảo quyền lợi, độ an toàn và được bảo vệ chủ thể nghiên cứu, phù hợp với nguyên tắc của tuyên ngôn Helsinki và các dữ liệu nghiên cứu đảm bảo và có thể tin cậy. Yêu cầu trong thực hiện một nghiên cứu thử nghiệm lâm sàng ở châu Âu, gồm GCP và GMP và đồng thời có sự xét duyệt theo hướng dẫn thử nghiệm lâm sàng chung châu Âu (Clinical Trial Directive_Directive 2001/20/EC) và hướng dẫn về GCP Directive (2005/28/EC). Thử nghiệm lâm sàng liên quan hay nói đúng hơn là bao gồm cả áp dụng cáp phép trên thị trường và liên quan đến EEA tiến hành đi theo với GCP (hướng dẫn 2001/83/EC, phụ lục I) và theo sửa đổi bổ sung có hướng dẫn 2003/63/EC. Một số thông tin liên quan GCP khác có thể truy cập tại website của các cơ quan tổ chức như của Council for International Organizations of Medical Science (CIOMS) và World Medical Association (WMA) và tuyên ngôn Declaration of Helsinki. GCP_các sản phẩm thuốc trên thú y (Veterinary Medicinal Products)Đối với thử nghiệm lâm sàng các thuốc trong lĩnh vực thú y, châu Âu đã có VICH GCP (VICH là một chương trình có sự thông nhất giữa 3 bên [EU-Japan-USA] nhằm hài hòa các yêu cầu về kỹ thuật trong đăng ký các sản phẩm trong lĩnh vực thú y, viết tắt của cụm từ đầy đủ là International Cooperation on Harmonisation of Technical Requirements for Registration of Veterinary Medicinal Products; VICH chính thức ra mắt và hoạt động vào tháng 4 năm 1996), đã được chấp thuận bởi CVMP vào tháng 7 năm 2000 và có hiệu lực vào tháng 7 năm 2001. Trong đó bao gồm các hướng dẫn về thiết kế nghiên cứu và cách tiến hành tất cả các thử nghiệm lâm sàng liên quan đến sản phẩm dùng trong thú y ở các loài chủ đích. Liên quan đến các tiêu chuẩn, tiêu chí và yêu cầu giữa 3 bên thống nhất trong một hệ thống đảm bảo chất lượng sao cho đáng tin cậy được diễn tả theo mô hình trên. Hướng dẫn tất cả cá nhân và tổ chức liên quan đến thiết kế nghiên cứu, tiến hành nghiên cứu, giám sát, ghi chép, thanh kiểm tra, phân tích và báo cáo về các dữ liệu trong một thử nghiệm lâm sàng trên các loài chính và phải đảm bảo rằng các nghiên cứu như vậy tiến hành và trình bày theo nguyên tắc chuẩn của Good Clinical Practice (GCP). Tương tự, đánh giá các sản phẩm dược trên người, thì các sản phẩm của thú y cũng có liên đới theo hướng dẫn 2001/82/EC. Nhiệm vụ của chương trình thử nghiệm lâm sàngChương trình thử nghiệm lâm sàng là một trong những tiêu điểm của FDA đối với vấn đề GCP đang diễn ra nhiều nghiên cứu thử nghiệm lâm sàng trên người theo điều luật của FDA. Liên quan đến GCP, chương trình GCP gồm có: ·Cộng tác với các chính sách của Cơ quan quản lý dược và thực phẩm của Mỹ (US_FDA); ·Góp phần vào khâu chỉ đạo và hướng dẫn suốt quá trình tham gia để bảo vệ quyền lợi con người theo FDA trên các nghiên cứu y sinh học có liên quan đén con người; ·Cộng tác với chương trình giám sát nghiên cứu y sinh học của FDA (FDA's Bioresearch Monitoring program) với các khía cạnh liên quan nghiên cứu thử nghiệm lâm sàng, làm việc cùng với bộ phân ban hành pháp luật của cơ quan FDA (FDA's Office of Regulatory Affairs _ORA); ·Góp phần vào các hoạt động một cách hài hòa theo chuẩn của GCP quốc tế; ·Có kế hoạch và tiến hành đào tạo cũng như các chương trình tiếp theo; ·Đóng vài trò như một liên lạc viên với văn phòng bảo vệ con người trong nghiên cứu (Office for Human Research Protection_OHRP) và tác ban ngành khác cũng như các bên liên quan bên ngoài tham gia. Một ví dụ minh họa là thử nghiệm lâm sàng thuốc sốt rét mới Trước đây, khi khoa học kỹ thuật chưa phát triển một cách đầy đủ thì hầu hết các thuốc điều trị sốt rét hiệu quả của chúng ta không có thiết kế hoặc công thức thuốc hợp lý (ngoại trừ thuốc nhóm antifolates). Chẳng hạn, nhóm thuốc quinoleines (Chloroquine, Amodiaquine, Mefloquine và Quinine) và nhóm thuốc endoperoxides (Artemisinin và các dẫn suất) được điều chế không có hiểu biết thấu đáo rõ ràng về đích ngắm phân tử mà thuốc sẽ tấn công (molecular target) và không đảm bảo rằng những thuốc này sẽ giết ký sinh trùng hay không và nếu có sẽ theo cách nào. Bước vào thế kỷ 21, có một điều nổi bật và tin tưởng là các nhà khoa học đã xác định được điểm đích tác động, thiết kế công thức thuốc hợp lý sẽ là hướng đi đúng trong việc tạo ra các thuốc sốt rét thế hệ mới. Sự lạc quan này một lần nữa đã khẳng định khi chúng ta có những thành quả gần đây về dự án bộ gen của muỗi và bộ gen của loại ký sinh trùng sốt rét nguy hiểm nhất P.falciparum để có đủ dữ liệu mà đi thẳng đến mục đích thiết kế các thuốc mới. Chúng ta đã và đang tiếp cận các tiến bộ về khoa học đến mức nay dường như đang ở trong kỷ nguyên hậu di truyền “postgenomic era” và ở đó chúng ta có những kỹ thuật tiên tiến trên toàn cầu có khả năng điều tra tất cả những gen và protein bên trong hầu hết vi sinh vật. Mặc dù điều này rất đơn giản, song người ta đã tranh cãi rất nhiều về nó; bằng những hiểu biết sâu hơn về các tuơng tác phức tạp giữa những phân tử bên trong vi sinh vật dưới điều kiện được xác định (chu kỳ sinh học, phơi nhiễm thuốc, …) và so sánh những phân tử chính trong vật chủ và tác nhân gây bệnh, từ đó xác định các điểm đích cho thuốc tác động lên ký sinh trùng một cách chính xác nhất. Tuy nhiên, để đi đến đích của nghiên cứu không phải lúc nào cũng bằng phẳng mà luôn luôn tồn tại những thử thách cho các nhà khoa học rất lớn vì trong khi các nổ lực và phát minh của họ quá khiêm tốn trong việc quản lý các dữ liệu và có chiến lược phát triển điều chỉnh khả năng thuốc đi trúng đích của vi sinh vật. Thật cần thiết, khi ứng dụng các tiêu chuẩn nghiêm ngặt trong nghiên cứu đích tác động của thuốc là công nghệ sàng lọc cao cấp và xác định đầy đủ thông tin ở chúng. Đồng ý rằng ta có nhiều cơ hội để thiết kế thuốc hợp lý trong thời đại này; song, sự đột phá đó còn ít, quy trình nghiên cứu thuốc quá đắt, nguy cơ thất bại cao. Chi phí để đưa ra một thuốc sốt rét mới đòi hỏi phải có sự tận tâm, tận lực và hiện diện một cộng đồng vùng sốt rét, nền công nghiệp dược vào cuộc, nên chi phí đầu tư không phải là nhỏ. Chi phí kinh tế trong nghiên cứu thuốc mới Dù thế nào đi chăng nữa các phương thức được dùng xác định thuốc sốt rét có tiềm năng và hiệu lực trên in vitro, rồi đến các bước quy đổi thành phần hoặc hợp chất phải phát triển thành thuốc, thì trong suốt tiến trình, lợi ích và nguy cơ bắt đầu được đánh giá. Mặc dầu những ý tưởng ban đầu làm cơ sở cho bước lập dự án thường bắt nguồn từ lý luận và chuẩn y, hầu hết các thuốc được phát triển nhờ vào công nghệ dược phẩm (phần lớn tập trung tại các quốc gia công nghiệp) ở mức đầu tư tài chính cao. Do vậy, mặc dù nhóm nghiên cứu và đội ngũ năng động trong y họa và khoa học thì tài chính bỏ ra vẫn mong đợi một lợi nhuận thuốc sốt rét phải mang tính cạnh tranh với thuốc cũ theo phân loại thuốc. Trong suốt quy trình làm, kết quả là nhiều hợp chất khác của chúng được khám phát vượt ngoài dự kiến Tuy nhiên, tạo ra các hợp chất này để được đăng ký thì lại cần các chuyên gia dược và mất một khoảng chi phí khác. Sự tận tâm tận lực như thế thì các công ty dược thường nhận được kết quả mỹ mãn giữa chi phí và hiệu quả của các quốc gia và tổ chức quốc tế ủng hộ. Sự đóng góp từ tổ chức đầu tư vốn để kinh doanh thuốc sốt rét rất hoan nghênh khi nghiên cứu về thuốc mới. vì ở đó, họ còn mong đợi đạt nhiều thành quả từ các nghiên cứu để tác động làm giảm tỷ lệ bệnh tật và tử vong do sốt rét. Thế nhưng, thực tế cho thấy những bước phát triển thuốc sốt rétquá chậm trong khi tình trạng kháng thuốc sốt rét đang chầm chậm đe dọa mạng sống nhiều người. Vậy, điều tối cần thiết và quan trong thời gian này là chúng ta phối hợp cùng nhau để chế ra thuốc mới, gồm cả phối hợp dược công lẫn tư. Các giai đoạn phát triển nghiên cứu thuốc mới Nói chung, hiệu lực và tính an toàn là tiêu chuẩn hàng đầu cho mọi thuốc mới trước khi đưa ra thị trường phải được thử ngiệm đầy đủ và hoàn toàn mang tính khoa học, ngay cả một số thuốc cũ nhưng dùng cho một số chỉ định mới. Do đó, điều lệ và quy định của xây dựng và phát triển thuốc mới rất nghiêm ngặt và chặt chẽ vì một “dấu ấn” trong quá khứ (ví như vụ thalidomide gây bao nhiêu biến dạng sau một thoèi gian sử dụng thuốc kéo dài). Ngày nay, nhu cầu công chúng là mong có thuốc chuẩn và độ an toàn cao ở mọi nơi: Mong đợi chính đáng là những chính quyền đưa ra điều luật như Cục Quản lý dược thực phẩm Mỹ (Food and Drug Administration-FDA), Tổ chức luật về sản phẩm và thuốc trong chăm sóc sức khỏe (Medicines and Healthcare Products Regulatory Agency -MHRA), Tổ chức đánh giá sản phẩm dược châu Âu (European Agency for the Evaluation of Medicinal Products-EMEA). Một lẽtự nhiên, thuốc được sử dụng trong vùng nhiệt đới phải có cùng chuẩn, hầu hết các quốc gia có điều luật riêng nhưng thường dựa trên quy trình đánh giá của các tổ chức FDA, MHRA, EMEA, hoặc các tổ chức tương đương. Các nhà sản xuất thuốc mới rất trân trọng chất lượng, độ an toàn và hiệu quả của một sản phẩm. Quy trình nghiên cứu phát triển có hồ sơ điều luật mất 10 năm, chi phí khoảng 100 triệu đo la Mỹ (USD). Một nền tảng để chấp nhận tất cả các giai đoạn phát triển thuốc mớiphải được hỗ trợbằng phân tích thống kê, số liệu thô phải được phân tích sẵn để xem lại và dữ liệu liệt kê phải kiểm tra với nguồn số liệu (số liệu về thử nghiệm hoặc hồ sơ lâm sàng). Những thông số theo dõi phải rất tóm tắt, đơn giản khi mô tả các giai đoạn chính của quá trình nghiên cứu, trên thực tế là nhiều giai đoạn tách bạch ra nhưng lại tiến hành song song nhau. Phát triển dược phẩm là ngay từ đầu sản xuất dược một số lượng lớncác hợp chất (với công đoạn hình thành công thức cho thuốc uống, thuốc đặt hậu môn, thuốc cho bằng đường ngoài ruột như tiêm truyền). Điều luật thực hiện GMP của quốc tế thường được dùng để đảm bảo rằng thuốc được chế ra phù hợp với tiêu chuẩn chấp nhận về chất lượng. Thực chất, những điều lệ này cần biết thành phần hóa học của thuốc (bao gồm xác định cả chất nhiễm) và đặc tính của thuốc đó (chẳng hạn độ tan rã). Chúng cần phải đảm bảo rằng (i) Thuốc phải ổn định về mặt lý hóa và (ii) Chất lượng đáng tin cậy. Đối với việc nghiên cứu để cho ra và được công nhận một thuốc mới nói chung và thuốc điều trị sốt rét nói riêng là một quá trình lâu dài, đáp ứng nhiều quy định hết sức nghiêm ngặt. Tại sao phải như vậy? Bởi vì thuốc được chấp nhận dùng trong điều trị, phòng bệnh và chẩn đoán phải đạt hai tiêu chuẩn: hiệu quả và an toàn. Tức là thuốc phải có tác dụng thực sự và trong quá trình sử dụng không gây tai biến quá nghiêm trọng cho người sử dụng. Chúng ta thường nghe có thuốc rất nổi tiếng, dùng lâu năm nhưng rồi bị cấm lưu hành chính vì đã được nghiên cứu, thử nghiệm lâm sàng rất bài bản song người ta vẫn không phát hiện các tác dụng phụ nghiêm trọng mà phải sau một thời gian sử dụng, số người sử dụng tăng đến số lượng đủ để tác dụng phụ gây chết người ấy lộ ra. Nghiên cứu công phu như thế và thử nghiệm lâm sàng như vậy song vẫn không lường được tính không an toàn của thuốc, nói chi đến những thuốc không được thử nghiệm, hay thử nghiệm không đúng các bước yêu cầu. Tại các nước tiên tiến, một dược phẩm chỉ được chấp nhận lưu hành trên thị trường khi đã theo đúng một quy trình nghiên cứu, và hiện nay tiến trình này đã được Công ước Quốc tế xem như khuôn mẫu để thuốc thực sự mang tính hiệu quả và an toàn. Tiến trình này thường cần có thời gian thường là từ 10-15 năm, thực hiện qua các giai đoạn: Thử nghiệm tiền lâm sàng-chưa thử can thiệp trên người Mục đích của thử nghiệm tiền lâm sàng là xác định cấu trúc hoá học, tính chất lý hoá của thuốc mới (nếu là bài thuốc thì các vị thuốc cũng phải được xác định các thành phần một cách rõ ràng); thử tác dụng dược lý, độc tính trên súc vật; xác định các tiêu chuẩn của các dạng bào chế nếu thuốc được dùng ở một dạng bào chế nhất định nào đó và khi thuốc đã thử trên súc vật chứng tỏ mức sơ bộ, người ta tính hiệu quả và an toàn, tác giả (cá nhân hay tập thể) mới xin hợp thức hoá bằng một bằng sáng chế và lập hồ sơ đệ trình lên cơ quan Nhà nước (chẳng hạn Cục quản lý dược Việt Nam hoặc FDA của Mỹ), trong bản nghiên cứu thử nghiệm thuốc mới ghi rõ các dữ kiện thử nghiệm tiền lâm sàng và tiến trình thử nghiệm trên người. Khi bản nghiên cứu thử nghiệm được chấp nhận, thì thuốc mới được phép thử nghiệm lâm sàng. Trước khi một thuốc dự định nghiên cứu trên người, phải mất rất nhiều thời gian để kiểm tra độc tính của nó trên động vật. Các điều luật về thực hành phòng thí nghiệm tốt (GLP-Good Laboratory Practice regulations) đảm bảo các dữ liệu phải rõ ràng trong hồ sơ. Những nghiên cứu bao gồm (i) nghiên cứu về mô hình tác động của thuốc và dược lý học một cách chi tiết; (ii) những nghiên cứu về độc tính của thuốc ở liều đơn. điều này sẽ giúp chúng ta phác thảo những nét chính khi quá liều; (iii) những nghiên cứu về độc tính đối với liều lặp lại trong hơn một loài (điều này có tác dụng là cung cấp thông tin hơn là thử nghiệm liều đơn) vì thường giúp ta mô tả những đặc điểm về nhiễm độc mạn tính; thử nghiệm về mô bệnh học và quan sát các hành vi một cách chi tiết và chặt chẽ trên động vật, thường là đặc điểm chìa khóa cho cả những nghiên cứu liều đơn và liều lặp lại); (iv) nghiên cứu tái sản xuất cho cả hai giới, kiểm tra tính ảnh hưởng lên khả năng sinh sản, kiểm tra tác dụng đảo ngược của bất kỳ tác dụng phụ nào, bằng chứng độc phôi thai và sinh quái thai và (v) các nghiên cứu sinh đột biến, thường đòi hỏi (trong nhiều trường hợp dữ liệu sinh ung thư cũng phải đề cập). Nghiên cứu trên người pha I Mục đích của pha I này là thuốc mới được thử nghiệm trên khoảng từ 20-100 người tình nguyện trưởng thành khoẻ mạnh (tức không phải là người bệnh) và nhằm biết tính chất dược động học của thuốc (cơ thể hấp thu, phân bố, chuyển hoá và bài tiết như thế nào) và độ an toàn, tức là xem thuốc có gây tác dụng phụ nào trên người hay không? Tất cả những pha nghiên cứu trên người phải chiếu theo các quy định về thực hành lâm sàng tốt (GCP-good clinical practice). Pha I có thể tiến hành trên những người trưởng thành khỏe mạnh (thường cả hai giới) hoặc những bệnh nhân mắc bệnh (dựa trên độc tính tiềm tàng của thuốc). Tổng số người nghiên cứu thường dưới 100. Mục đích chính thường là (i) điều tra tính an toàn (dựa trên các thông số sinh lý và thử nghiệm trên chủ thể (chẳng hạn ECG) và tính dung nạp (thường dựa vào các đánh giá trên người tình nguyện), với các liều khác nhau và (ii) nghiên cứu về dược động học, các số liệu phải được so sánh và đối chiếu với số liệu quan sát trên động vật. Nghiên cứu trên người pha II Giai đoạn này nhằm đánh giá thuốc mới được thử trên số lượng người bệnh (tức là không phải người khoẻ) khoảng 100-300, để xem thuốc có hiệu quả và an toàn không. Để kết quả của cuộc thử nghiệm mang tính chính xác, khoa học và kết quả nghiên cứu phải dựa vào thực nghiệm khoa học. Phương pháp mù đôi ngẫu nhiên thường đựợc các nhà nghiên cứu áp dụng nhiều vì cả bệnh nhân và thầy thuốc điều trị không biết thuốc sử dụng là thuốc thật hay thuốc giả dược (placebo) để việc thử nghiệm mang tính khách quan và khoa học. Nghiên cứu pha này liên quan đến đánh giá thuốc mới có đánh dấu trong một nghiên cứu mở trên bệnh nhân. Trong nghiên cứu Pha II các bước phải được thực hiện một cách chi tiết bằng những nhà lâm sàng kinh nghiệm với bệnh đang được điều trị. Một trong những yếu tố chính của pha II là mô tả liên quan giữa liều thuốc và hiệu lực thuốc. Các nghiên cứu chuyên biệt này đòi hỏi nhóm đối tượng đặc biệt như là bệnh nhân với khoảng tuổi cách xa và bệnh nhân bị tổn thương gan, thận.Thường có một yếu tố về dược động học cho nghiên cứu pha II này và khía cạnh an toàn và tính dung nạp thuốc là rất quan trọng. Nghiên cứu tương tác thuốc giữa thuốc mới với các thuốc được chỉ định đồng thời cũng là phần quan trọng của pha II. Vài trăm bệnh nhân được sử dụng trong nghiên cứu pha này. Nghiên cứu trên người pha III Thuốc mới được thử trên số lượng lớn người bệnh nội trú ở nhiều bệnh viện / trung tâm y tế (multicenter) khác nhau và cả bệnh nhân ngoại trú (có thể trên 10.000 người nếu có thể) mục đích là đúc kết rằng thuốc mới có tính hiệu quả và an toàn trên người bệnh. Chính nhờ thử nghiệm trên một số lượng lớn người bệnh như thế mà các tác dụng phụ và tác dụng ngoạị ý xuất hiện tần xuất thấp có khả năng phát hiện được nhiều hơn và đánh giá rút kinh nghiệm chuẩn hơn. Nếu pha III này đạt yêu cầu, hãng dược phẩm sẽ nộp lên các cơ quan quản lý có chức năng tất cả hồ sơ theo yêu cầu để được xét duyệt trước khi đưa ra giai đoạn thị trường. Thông thường ở các nước tiên tiến như Mỹ, Singapore, Pháp, Italia, Nhật,...thời gian để xét cho phép lưu hành thuốc mới thường từ 1-2 năm. Những nghiên cứu pha III trên người là những TNLS ngẫu nghiên và rất quy mô, trong đó hiệu lực, độ an toàn và tính dung nạp thuốc mới được so sánh với nhưng thông số như trên nhưng thuốc giả dược hoặc thuốc chuẩn. Trong nghiên cứu thuốc sốt rét, sử dụng giả dược rõ ràng không thể được và vì thế phải so sánh thuốc mới với các thuốc chuẩn/ điều trị chuẩn theo các thông số. Số lượng bệnh nhân trong nghiên cứu pha III thường 1.000-2.000. Do đó, trong hầu hết các trường hợp, hồ sơ của thuốc mới lại không biết một cách hoàn hảo ở thời điểm bắt đầu nghiên cứu. Ngoài ra, dữ liệu về độ an toàn được đo bằng TNLS ngẫu nhiên, nên kết quả có thể gần giống với thực tế. Sự chấp nhận dùng thuốc của bệnh nhân thường được đánh giá, giám sát một cách chặt chẽ trong pha III này. Ngược lại, dưới những điều kiện bình thường, bệnh nhân thường bất hợp tác hoặc hài lòng ít với việc dùng chế độ điều trị nhất là nếu khi họ có biến chứng hoặc tai biến. Pha III thường chọn quần thể bệnh nhân rất chuẩn, quần thể nghiên cuứ đồng nhất và bênh nhân phải tuân thủ điều kiện nghiêm ngặt. Pha III thường có một giai đoạn theo dõi ngắn. Do vậy, nếu thuốc tác động bị mất đi theo thời gian thì một pha III dễ bị thất bại và cần xem xét thêm. Nghiên cứu trên người pha IV Giai đoạn này còn gọi là pha “hậu mãi”, nghĩa là thuốc đã chấp nhận bán ra thị trường nhưng phải được tiếp tục theo dõi về độ an toàn và các tác dụng phụ. Giai đoạn này thường trên 10 năm và do chính hãng dược phẩm cho ra đời thuốc đó thực hiện. Quy trình lâu dài và theo dõi nghiêm túc nhưng gần đây cũng có vài ngoại lệ (một số thuốc chưa đủ quy trình thử nghiệm đã tung ra thị trường); Một số thuốc do nhu cầu tính bức thiết của nó như thuốc điều trị HIV thì thông thường thời gian TNLS có thể rút ngắn hoặc một số mỹ dược phẩm không đảm bảo chất lượng và hiệu quả trước khi đưa ra thị trường như men đã được sự đồng ý của cơ quan FDA của Mỹ, Cục an toàn dược phẩm của Pháp (Afssaps) hoặc Cục quản lý dược Việt Nam. Hồ sơ quy định sẽ phải thành lập cho thuốc mới đòi hỏi tiêu chuẩn về chất lượng và khía cạnh hiệu lực và tính dung nạp thuốc; Hồ sơ về độ an toàn thuốc phải được ghi chép cẩn thận và xem xét trên khoảng 2.000 bệnh nhân. Đây chính là những thông tin không đầy đủ mà từ các người có thẩm quyền của quốc gia có thể đặt quyết định tiếp nối thuốc sốt rét mới vào trong chính sách, bởi vì điều kiện hoạt động sẽ được kích thích và đi sâu nghiên cứu con đưa ra quyết định và nghiên cứu. Vấn đề quan tâm chính khi đánh giá quy trình sử dụng thuốc mới ở châu Phi đặt ra là có các tiêu chuẩn về độ an toàn, sự chấp nhận, hiệu quả và tính kháng thuốc. Điều đáng quan tâm ở điểm mà thuốc mới sẽ được tung ra thị trường như là một thuốc chỉ dánh cho kê đơn, nghĩa là sử dụng dưới sự giám sát chuyên biệt trong một hệ thống y tế chính quy bài bản. (i) Độ an toàn. Hầu hết các hồ sơ về điều luật về bất kỳ bệnh gì, không chỉ riêng sốt rét thì số liệu nghiên cứu sẽ ít hơn 2.000 người được cơ hội tiếp nhận thử thuốc mới. Tuy nhiên, tỷ lệ bệnh nhân bị đe dọa mạng sống nhưng đặc ứng cấp thường thấp hơn 1/10.000. Do đó, sẽ ít có cơ hội mô tả hồ ớ AE của một thuốc mới một cách đầy đủ cho đến khi đưa ra thì trường. Những hệ thống cảnh báo thận trọng về dược đã được thiết lậptại các quốc gia công nghiệp, như là hệ thống card vàng “yellow card system” ở Vương quốc Anh. Song, những hệ thống dược cảnh giác còn ít ở châu Phi và đạt được chất lượng cao nhưng số liệu hậu thị trường mới là những thử thách cho họ. (ii) Sự tôn trọng tối ưu Chỉ một tỷ lệ bênh nhân bất kỳ nơi đâu trên thế giới phải được giám sát và tôn trọng họ khi dùng phác đồ điều trị thậm chí khi cần thiết phải giải thích một cách đầy đủ, cẩn thận. Đối với một vài thuốc, sự hài lòng của bệnh nhân không trọn vẹn không thể ảnh hưởng đến hoàn toàn két quả điều trị nhưng với hầu hết các thuốc sốt rét, sự quản lý chặt chẽ dùng thuốc là rất quan trọng. Một số yếu tố được cho là không phụ thuộc một cách tương đối trong một số tình huống sử dụng thuốc (ví dụ ở nơi có đơn vị chăm sóc y tế bài bản và không bài bản). (i) Về kinh nghiệm thì các vấn đề có thể mong đạt được từ chế độ điều trị là có thể kéo dài, mở rộng ra (muộn hơn 3 ngày), khi đó liều cũng đòi hỏi là hơn 1 ngày khi đó cần thiết điều trị với công thức thuốc cũng hơn 2. (ii) Hầu hết các bênh nhân mắc sốt rét rất nghèo và chi phí thuốc đã ảnh hưởng lên quá trình dùng thuốc; nên chế độ điều trị có thể hoãn lại khi số tiền của họ quá nhỏ. (iii) Công thức thuốc không phù hợp cũng có thể là một rào cản. Có lẽ bằng quan sát trực quan, liều thuốc dạng dung dịch của trẻ em châu Phi ít có chính xác so với khi dùng dạng thuốc viên. Điều này phản ánh kinh nghiệm khó thực hiện vì nhiều bà mẹ ước tính lượng thuốc trước khi cho uống. (iv) Nếu một thuốc thường gây ra tác dụng phụ (chloroquine gây ngứa hoặc mefloquine gây buồn nôn) thì việc bệnh nhân phối hợp để điều trị với các thuốc như thế sẽ khó khăn. (v) Trước khi đóng gói thuốc vào trong một đơn vị liều thì phải tính toán đến sự phối hợp này, đặc biệt ở châu Phi. Ngoài ra, cần lưu ý đến thiết kế mẫu mã của tờ rơi có thông tin (sẽ giúp rất nhiều trong việc nâng cao tính phối hợp này. Tuy nhiên, ngay cả những thuốc đạt ưu điểm nhất (liều dùng đơn giản, chi phí thấp, độc tính ít, công thức thuốc phù hợp và liều trong một đơn vị đóng gói cho phép) vẫn còn phạm vi rất lớn để nói khi sử dụng không đúng cách như trong công việc kê đơn, pha chế phân phối và bệnh nhân dùng thuốc. Phạm vi này không được nghiên cứuthấu đáo mãi đến gần đây, nhưng có một sự chấp nhận về giáo dục thông tin một cách bài bản và công tác truyền thông rõ ràng làm thế nào để phổ cập kiến thức dùng thuốc tốt nhất, an toàn nhất. Những công việc như thế sẽ rất chi tiết và lệ thuộc rất nhiều vào cơ sở y tế có làm đúng bài bản không. Hiệu quả điều trị và sự kháng thuốc. trong các thử nghiệm ngẫu nhiên, hoàn toàn các bộ phận lâm sàng nghiên cứu về hồ sơ thuốc thật cẩn thận khi lựa chọn bệnh nhân uống thuốc dưới sự giám sát. Đây là những tình huống tự giác rất cao ở châu Phi; thậm chí các nơ chăm sóc y tế, hầu như điều trị sốt rét liên quan đến bệnh nhân ngoại trú. Nên việc theo dõi diễn tiến bệnh quả thật khó khăn liệu bênh nhân dùng thuốc theo ý không. Khi dùng cùng với một thuốc chống nhiễm khuẩn khác thì hiệu dĩ nhiên hiệu quả thuốc sẽ khonog đạt được kết quả tối ưu. Những khó khăn trong phối hợp thuốc sốt rét Nếu thuốc được cấp giấy phép trước đây thì các thuốc sử dụng có thể phối hợp nhau (như thuốc phối hợp artemisinin- ACTs), có hay không việc cùng đóng gói và cùng pha ché công thức phải theo một quy định. Trong tâm chính khi phối hợp thuốc là xem hiệu quả mang lại cho người bệnh phải lớn hơn nhiều so với nguy cơ. Nếu các thuốc có đặc tính có thể cộng lực nhau (như thuốc Sulfadoxine/ Pyrimethamin) thì trường hợp này có thể liên quan đến quy trình làm, sử dụng các thuốc riêng biệt Pyr & Sul sẽ tính đến khâu hiệu quả (nhất là thuốc nhóm sulfonamide). Ngoài ra, các quy định đòi hỏi tính ổn định trong các chế phẩm kết hợp và và hiệu quả cũng như độc tính tiền lâm sàng và đặc tính dược lý, bao gồm cả dược động học của thuốc đó. Đạo đức trong nghiên cứu y sinh học Lịch sử phát triển: Đạo đức trong nghiên cứu y sinh học là các nguyên tắc, các chuẩn mực áp dụng trong nghiên cứu đặc biệt là các nghiên cứu y sinh học liên quan đến con người. Có 3 nguyên tắc cơ bản của đạo đức trong nghiên cứu y sinh học, đó là: 1.Từ tâm (beneficence), điều này đòi hỏi kết quả phải là điều tốt lành, tránh điều có hại hoặc lợi ích phải vượt xa các nguy cơ hoặc điều có hại; 2.Tôn trọng quyền cá nhân (respect for rights), bao gồm quyền tự lựa chọn của đối tượng tham gia nghiên cứu và bảo vệ những đối tượng không có khả năng tự quyết; 3.Sự công bằng (justice), điều này đòi hỏi bảo đảm phân bố đều về trách nhiệm và lợi ích của đối tượng tham gia nghiên cứu, nhà khoa học, nhà sản xuất, cá nhân cũng như của xã hội. Những nguyên tắc về đạo đức trong nghiên cứu y sinh học đặc biệt là những nguyên tắc đạo đức trong thực hành y dược, được đề cập từ rất sớm, nhất là các thử nghiệm phương pháp chữa bệnh hoặc chẩn đoán mới. Văn kiện quốc tế đầu tiên về đạo đức trong nghiên cứu là điều lệ Nuremberg 1947. Sau điều lệ Nuremberg, năm 1948 Hội đồng Liên hợp quốc đã thông qua tuyên bố toàn cầu về Quyền con người, Hội đồng Liên hợp quốc cũng đã thông qua Hiệp ước quốc tế về Quyền công dân và chính trị vào năm 1966. Năm 1964, Hiệp hội Y học thế giới (World Medical Association - WMA) đã ra tuyên ngôn Helsinki, đó là một văn bản cơ sở trong lĩnh vực đạo đức nghiên cứu y sinh và ảnh hưởng tới việc hình thành hệ thống luật pháp của quốc gia, khu vực và quốc tế. Tuyên bố này được chỉnh lý nhiều lần, lần cuối cùng vào năm 2000, là một lời tuyên bố quy mô toàn cầu về đạo đức trong nghiên cứu liên quan đến con người. Nó giúp cho việc hình thành những hướng dẫn về đạo đức cho các bác sĩ, các nhà khoa học tham gia vào các nghiên cứu y sinh lâm sàng và cận lâm sàng có liên quan đến con người là đối tượng nghiên cứu. Từ điều lệ Nuremberg 1947 đến tuyên ngôn Helsinki 1964 rồi đến các hướng dẫn của Hội đồng các tổ chức quóc tế về khoa học y học (CIOMS_Council for International Organizations of Medical Sciences) năm 1982, các tài liệu về đạo đức nghiên cứu trong các nghiên cứu y sinh học có liên quan đến con người được hoàn thiện dần thông qua các lần chỉnh sửa. Tuyên ngôn Helsinski được chỉnh sửa năm 1975 sau đó được rà soát và bổ sung vào các năm 1980, 1983, 1989, 1996 và năm 2000. Ngày nay, vấn đề đạo đức trong nghiên cứu y sinh học đã có các văn bản hướng dẫn quốc tế giúp mỗi quốc gia trên cơ sở đó xây dựng quy định và hướng dẫn thực hiện cụ thể.Các chuẩn mực trong nghiên cứu y sinh học bao gồm những nội dung cơ bản sau đây: 1.Nghiên cứu y sinh phải tuân theo các nguyên tắc khoa học và phải dựa trên nghiên cứu trong phòng thí nghiệm và trên động vật một cách đầy đủ, và phải dựa trên các kiến thức thấu đáo từ các tài liệu khoa học; 2.Thiết kế từng phép thử nghiệm trên đối tượng con người phải được hình thành trong đề cương nghiên cứu và phải được đánh giá bởi hội đồng độc lập; 3.Nghiên cứu thử nghiệm phải được thực hiện bởi cán bộ có đủ trình độ khoa học tương xứng và được giám sát bởi các chuyên gia y học có kinh nghiệm lâm sàng; 4.Bất cứ nghiên cứu y sinh học nào có đối tượng nghiên cứu là con người cũng cần phải được đánh giá cẩn thận các nguy cơ có thể lường trước so với các lợi ích có thể đạt được cho đối tượng nghiên cứu và các đối tượng khác. Quan tâm đến lợi ích của đối tượng nghiên cứu luôn phải đặt trên lợi ích của khoa học và của xã hội; 5.Quyền của đối tượng nghiên cứu được bảo đảm về sự toàn vẹn luôn luôn phải được đặt lên hàng đầu. Tất cả các điều dự phòng phải được tiến hành để bảo đảm sự bí mật riêng tư của đối tượng và hạn chế tác động của nghiên cứu lên sự toàn vẹn về thể chất và tâm thần của đối tượng nghiên cứu và lên nhân phẩm của đối tượng; 6.Sự chính xác của các kết quả nghiên cứu phải được bảo vệ; 7.Bất cứ một nghiên cứu nào tiến hành trên con người, mỗi một đối tượng dự kiến tham gia nghiên cứu phải được biết thông tin đầy đủ về mục tiêu, các phương pháp, các lợi ích có thể và các tác hại có thể gây ra cho họ trong nghiên cứu, cũng như những phiền muộn có thể gây ra; 8.Khi đạt được sự chấp thuận tham gia trong nghiên cứu sau khi có được thông tin của đối tượng tham gia nghiên cứu, bác sĩ phải đặc biệt thận trọng nếu đối tượng trong tình trạng phụ thuộc vào bác sĩ. Không được gây áp lực hoặc đe doạ bắt buộc đối tượng tham gia nghiên cứu; 9.Trong trường hợp đối tượng thiếu hành vi năng lực, việc thông tin phải đạt được từ người có trách nhiệm pháp lý phù hợp theo luật pháp của quốc gia; 10.Các đối tượng tham gia nghiên cứu được rút khỏi nghiên cứu bất cứ lúc nào. Hội đồng đạo đức trong nghiên cứu y sinh học Hội đồng Đạo đức trong nghiên cứu y sinh học (còn được gọi là Hội đồng đạo đức độc lập - Independent Ethics Committee-IEC) là một hội đồng ở cấp địa phương (cấp cơ sở), cấp quốc gia hoặc cấp liên quốc gia được thành lập bao gồm các thành viên là các nhà khoa học, các chuyên gia về y tế và các thành viên có thể không thuộc ngành y. Hội đồng có nhiệm vụ xét duyệt các nghiên cứu thử nghiệm lâm sàng (TNLS), đưa ra các ý kiến chấp thuận hoặc không chấp thuận đối với TNLS nhằm bảo đảm sự an toàn, quyền lợi, sức khỏe của đối tượng tham gia nghiên cứu, đảm bảo tính công khai, khoa học và minh bạch trong việc xét duyệt. Tại Việt Nam, năm 2008 Bộ trưởng Bộ Y tế đã ký Quyết định số 661/QĐ-BYT ngày 27/2/2008 về việc thành lập Hội đồng Đạo đức trong nghiên cứu y sinh học - Bộ Y tế nhiệm kỳ 2008 - 2012. Nhiệm vụ chính của Hội đồng Đạo đức trong nghiên cứu y sinh học là đảm bảo quyền lợi, sự an toàn và tình nguyện tham gia của những đối tượng tham gia nghiên cứu, bảo đảm sự công bằng đối với tất cả các bên tham gia nghiên cứu, bảo đảm tính khoa học, khả thi của nghiên cứu, sự an toàn cho nghiên cứu viên và cộng đồng. Thẩm định, xét duyệt hồ sơ nghiên cứu y sinh học (đề cương nghiên cứu, các báo cáo và tài liệu có liên quan) bảo đảm tính pháp lý, khách quan, trung thực. Theo dõi, kiểm tra, giám sát việc tuân thủ nghiên cứu theo tiêu chuẩn thực hành lâm sàng tốt. Đánh giá thẩm định các kết quả nghiên cứu theo đề cương nghiên cứu đã được phê duyệt trên cơ sở các hướng dẫn và quy định hiện hành. Tập huấn, hướng dẫn và phát triển đội ngũ nghiên cứu viên cho ngành y tế theo các tiêu chí về Thực hành lâm sàng tốt (GCP) và đạo đức trong nghiên cứu. Ngành y tế Việt Nam hội nhập với quốc tế trong lĩnh vực nghiên cứu TNLS và đạo đức trong nghiên cứu Trong xu thế hội nhập với thế giới cũng như tôn trọng các nguyên tắc cơ bản về thực hành tốt thử nghiệm lâm sàng, đạo đức trong nghiên cứu và tôn trọng quyền con người, trong những năm qua ngành y tế Việt Nam đã đạt được một số kết quả bước đầu trong việc xây dựng, thiết lập và phát triển mạng lưới thực hành lâm sàng tốt của mình. Thể hiện với sự phát triển và hội nhập đó là, năm 2008 Bộ trưởng Bộ Y tế đã ký Quyết định số 661/QĐ-BYT ngày 27/2/2008 về việc thành lập Hội đồng Đạo đức trong nghiên cứu y sinh học - Bộ Y tế nhiệm kỳ 2008 - 2012. Ngày 7/3/2008 Bộ trưởng Bộ Y tế đã ký Quyết định số 799/QĐ-BYT về việc ban hành Hướng dẫn thực hành tốt thử nghiệm lâm sàng thuốc nhằm chuẩn hóa quy trình triển khai nghiên cứu TNLS thuốc tại Việt Nam phù hợp với các hướng dẫn của quốc tế cũng như tuân thủ luật pháp của Việt Nam. Đây là một Quyết định nhằm hướng dẫn cho nhà nghiên cứu, nhà tài trợ, các cơ quan quản lý, các hội đồng xét duyệt về đạo đức và khoa học trong thiết kế, tiến hành, thực hiện, theo dõi, giám sát, kiểm định, ghi chép, phân tích và báo cáo đối với các TNLS. Nó đảm bảo độ tin cậy, tính chính xác của các dữ liệu được báo cáo và sự chấp nhận mang tính quốc tế đối với các kết quả TNLS, đồng thời bảo đảm sự an toàn, quyền của các đối tượng nghiên cứu trong các TNLS. Bên cạnh việc ban hành các văn bản, hướng dẫn, quy chế về thực hành tốt TNLS và đạo đức trong nghiên cứu, trong thời gian vừa qua, Bộ Y tế còn tổ chức các khóa đào tạo về GCP và đạo đức trong nghiên cứu cho các nghiên cứu viên, các nhà khoa học, các nhà sản xuất nhằm cập nhật kiến thức và chuẩn hóa các nội dung về thực hành tốt thử nghiệm lâm sàng thuốc tại Việt Nam. Cùng với việc đào tạo cán bộ và các nghiên cứu viên lâm sàng, Bộ Y tế đã xây dựng Dự án phát triển và chuẩn hoá các đơn vị nghiên cứu thử nghiệm lâm sàng tốt theo tiêu chí về GCP. Trong thời gian tới, Bộ Y tế sẽ ban hành tiêu chuẩn Đơn vị đủ điều kiện thực hành lâm sàng tốt (Standard for GCP Unit) làm cơ sở xem xét, thẩm định và công nhận các bệnh viện/ viện nghiên cứu đủ điều kiện để triển khai nghiên cứu thử nghiệm lâm sàng thuốc mới, sản phẩm mới (CRU: Clinical Research Units). Với mục tiêu phát triển lâu dài và bền vững trong lĩnh vực nghiên cứu lâm sàng, trong giai đoạn tới Bộ Y tế sẽ thiết lập các dự án và kết hợp cùng các tổ chức quốc tế, Chính phủ của các quốc gia nhằm xây dựng Trung tâm nghiên cứu Thử nghiệm lâm sàng (Clinical Research Central) đạt theo chuẩn quốc tế cũng như thành lập Văn phòng Quốc gia về nghiên cứu lâm sàng (National Office for Clinical Research) đặt tại Bộ Y tế nhằm đưa công tác nghiên cứu lâm sàng tại Việt Nam theo chuẩn mực quốc tế. Đạt được một số kết quả ban đầu như trên là do sự chỉ đạo sát sao của lãnh đạo Bộ, sự năng động của Vụ Khoa học và đào tạo cũng như sự phối hợp, hỗ trợ của các chuyên gia trong nước, các chuyên gia quốc tế và các tổ chức quốc tế (US FDA, FHI, đại học Oxford, Sanofi, GlaxoSmithKline...). Quy trình chủ yếu trong quá trình thực hiện nghiên cứu TNLS thuốc tại Việt Nam Đăng ký nghiên cứu thử thuốc trên lâm sàng -Nhà tài trợ chuẩn bị hồ sơ đăng ký bao gồm: đơn đề nghị thử thuốc trên lâm sàng, đề xuất chủ nhiệm đề tài, cơ quan chủ trì (tổ chức nhận thử) kèm theo hồ sơ chứng minh về sản phẩm. -Gửi hồ sơ nói trên về Bộ Y tế (Vụ Khoa học và Đào tạo). Trong vòng 15 ngày làm việc kể từ khi nhận được hồ sơ theo đúng quy định, Bộ Y tế sẽ có văn bản trả lời làm cơ sở cho Nhà tài trợ triển khai các bước tiếp theo. Xây dựng hồ sơ nghiên cứu Căn cứ vào văn bản chấp thuận của Bộ Y tế, nhà tài trợ phối hợp với Chủ nhiệm đề tài xây dựng hồ sơ nghiên cứu thử thuốc trên lâm sàng bao gồm: -Nhà tài trợ cung cấp hồ sơ thông tin về sản phẩm, các căn cứ pháp lý, các nội dung đề xuất đề cương nghiên cứu cho chủ nhiệm đề tài và cơ quan chủ trì đề tài (tổ chức nhận thử thuốc trên lâm sàng). -Chủ nhiệm đề tài phối hợp cùng nhà tài trợ và các thành viên nhóm nghiên cứu thiết kế đề cương nghiên cứu, chuẩn bị đầy đủ các hồ sơ, đề cương, các văn bản pháp lý theo đúng các nội dung yêu cầu tại phần danh mục các tài liệu cần thiết tiến hành nghiên cứu thử nghiệm lâm sàng thuốc (bao gồm cả hợp đồng nghiên cứu giữa nhà tài trợ và cơ quan chủ trì - cơ quan nhận thử). Nộp hồ sơ nghiên cứu thử thuốc trên lâm sàng Hồ sơ nghiên cứu thử nghiệm lâm sàng phải được gửi về Bộ Y tế (Vụ Khoa học và Đào tạo) làm cơ sở cho việc thẩm định, xem xét và phê duyệt. Trong đó, trách nhiệm nộp hồ sơ của các bên như sau: -Nhà tài trợ chịu trách nhiệm nộp hồ sơ về sản phẩm đề xuất thử nghiệm cho cơ quan quản lý xem xét, thẩm định. -Chủ nhiệm đề tài và cơ quan chủ trì đề tài (tổ chức nhận thử thuốc trên lâm sàng) chịu trách nhiệm nộp hồ sơ đề cương nghiên cứu cho hội đồng đạo đức trong nghiên cứu thẩm định xét duyệt. -Thường trực của hội đồng đạo đức nghiên cứu và cơ quan quản lý nghiên cứu thử nghiệm trên lâm sàng đặt tại Vụ Khoa học và Đào tạo - Bộ Y tế. Chỉ có những hồ sơ nộp về Bộ Y tế trước ngày 20 hàng tháng (theo dấu công văn đến của phòng hành chính - Văn phòng Bộ) mới được xem xét thẩm định trong tháng đó. Những hồ sơ nộp sau thời hạn nói trên sẽ chuyển sang thẩm định ở tháng sau. Thẩm định, phê duyệt các nghiên cứu thử thuốc trên lâm sàng Thẩm định hồ sơ sản phẩm Trong vòng 30 ngày làm việc sau khi nhận đầy đủ hồ sơ đăng ký thử thuốc trên lâm sàng theo quy định, Bộ Y tế sẽ tổ chức họp ban thẩm định hồ sơ sản phẩm thử nghiệm lâm sàng theo các quy định hiện hành. Thẩm định hồ sơ nghiên cứu Trong vòng 30 ngày làm việc sau khi nhận đủ hồ sơ đăng ký thử thuốc trên lâm sàng theo quy định, song song với việc thẩm định hồ sơ sản phẩm, Bộ Y tế sẽ tổ chức họp hội đồng đạo đức trong nghiên cứu y sinh học theo các quy định hiện hành. Thông báo kết quả Trong vòng 15 ngày sau khi có các kết quả thẩm định của ban thẩm định hồ sơ sản phẩm và hội đồng đạo đức nghiên cứu, Vụ Khoa học và Đào tạo sẽ tổng hợp, hoàn chỉnh hồ sơ, biên bản và thông báo cho nhà tài trợ, cơ quan chủ trì đề tài (tổ chức nhận thử thuốc trên lâm sàng) bổ sung, hoàn chỉnh hồ sơ nghiên cứu thử thuốc trên lâm sàng (nếu có). Phê duyệt Trong vòng 15 ngày làm việc, Vụ Khoa học và Đào tạo sẽ tổng hợp, hoàn chỉnh các hồ sơ, đề cương nghiên cứu thử thuốc trên lâm sàng để trình bộ trưởng phê duyệt. Chỉ những hồ sơ được chấp thuận bởi cả hai khâu thẩm định nói trên (bao gồm: thẩm định hồ sơ sản phẩm, thẩm định hồ sơ nghiên cứu) mới được Bộ Y tế xem xét phê duyệt. Đối với những hồ sơ không được chấp thuận bởi cả hai khâu hoặc một trong hai khâu thẩm định nói trên, Bộ Y tế sẽ có văn bản thông báo tới nhà tài trợ và chủ nhiệm đề tài. Tài liệu tham khảo 1.Nguyễn Thị Kim Tiến.Thực hành tốt thử nghiệm lâm sàng và các vấn đề về đạo đức trong nghiên cứu y sinh học. Sức khỏe và đời sống 27/2/2009.2.Nguyễn Thị Kim Tiến.Thực hành tốt thử nghiệm lâm sàng và các vấn đề về đạo đức trong nghiên cứu y sinh học. Sức khỏe và đời sống 26/2/2009.3.Nguyễn Văn Tuấn. Y đức và Nghiên cứu y học. http://nhakhoathammy.com.vn 4.Huỳnh Hồng Quang. Nghiên cứu thuốc sốt rét mới & công thức phối hợp. http://www.impe-qn.org.vn 5.Council for International Organizations of Medical Sciences (CIOMS).International Ethical Guidelines for Biomedical Research Involving Human Subjects. Prepared by CIOMS in collaboration with the World Health Organization (WHO) 6.Joseph (2007). Ethical problems still surface in high data quality trials.(Ethics). BioResearch Compliance Report June 1, 2007 COPYRIGHT 2007 Washington Information Source, Inc. This material is published under license from the publisher through the Gale Group, Farmington Hills, Michigan. All inquiries regarding rights should be directed to the Gale Group. (Hide copyright information) 7.Hình ảnh trích từ trên các website của FDA, WHO, WHO- TDR, CIOMS,…
|