|
Làm thế nào thực hiện bản chấp thuận tham gia nghiên cứu sau khi thông hiểu về nghiên cứu
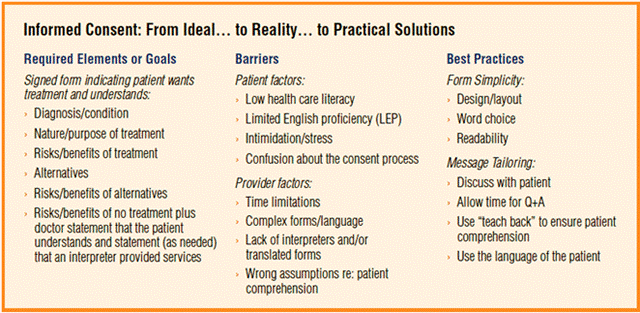
Điều quan trọng là cần hiểu rằng chấp thuận sau khi thông hiểu là một tiến trình bắt đầu bằng việc tuyển vào và sàng lọc một đối tượng và ký tên vào hồ sơ chấp thuận, sau đó tiến trình này sẽ tiếp tục trong suốt thời gian đối tượng tham gia cuộc nghiên cứu cũng như sau khi chấm dứt cuộc nghiên cứu. Tiến trình lấy chấp thuận tham gia nghiên cứu, nhất là thử nghiệm lâm sàng thuốc và vaccine, bao gồm: ·Các nỗ lực tuyển mộ bao gồm các phương pháp nâng cao nhận thức về vấn đề hoặc tạo mối quan hệ ban đầu, có thể được thực hiện qua nhiều cách như duyệt xét hồ sơ y khoa cho đến các quảng cáo và các tài liệu tuyển mộ khác . ·Cung cấp thông tin cụ thể và trả lời các câu hỏi về nghiên cứu cho các đối tượng theo cách dễ hiểu, đồng thời cho đối tượng có đủ thì giờ để cân nhắc việc tham gia. ·Giành được sự chấp thuận tự nguyện tham gia trong cuộc nghiên cứu từ đối tượng. Mặc dù đối tượng có thể đồng ý tham gia cuộc nghiên cứu, họ có thể rút ra vào bất cứ lúc nào. Việc chấp thuận mang tính chất liên tục, vì thế một phần của tiến trình chấp thuận là xác định rằng đối tượng vẫn muốn tham gia nghiên cứu. ·Lập ra kế hoạch cung cấp thông tin mới để chia sẻ với các đối tượng đã từng tham gia, thậm chí sau khi cuộc nghiên cứu đã chấm dứt. Có sự nhất trí về tầm quan trọng của việc chấp thuận sau khi thông hiểu. Chấp thuận sau khi thông hiểu là minh chứng cho thấy các nghiên cứu viên và những người có liên quan trong cuộc nghiên cứu trên đối tượng con người biểu lộ sự tôn trọng đối với đối tượng nghiên cứu, và cũng là điều bắt buộc bởi Sở Y Tế và Dịch Vụ Nhân Sinh (DHHS) tại 45 CFR 46 và Cơ Quan Quản lý Thực dược phẩm tại 21CFR 50. Các quy định đó được đưa ra để: ·Bảo vệ cho các đối tượng con người/những người tình nguyện; ·Đảm bảo rằng các đối tượng nghiên cứu tiềm năng hiểu rõ về các ích lợi và rủi ro có liên quan tới việc họ tham gia cuộc nghiên cứu; ·Cung cấp cho các đối tượng có thể sẽ tham gia nghiên cứu mọi thông tin cần thiết để đi đến quyết định có tham gia cuộc nghiên cứu hay không. Bộ Quy chế liên bang (CFR) được ấn hành trong Công báo liên bang, là một ấn bản của Chính phủ Liên bang nhằm hệ thống hóa các điều lệ chung và thường trực của các ban ngành và cơ quan thực thi. Có 50 chương tiêu biểu cho các lĩnh vực rộng lớn phải tuân thủ theo quy định của liên bang. CFR được cập nhật mỗi năm một lần và được phát hành hằng quý. Mục đích của môn học này là để cung cấp kiến thức cơ bản về sự chấp thuận sau khi thông hiểu và quy trình đat được sự chấp thuận sau khi thông hiểu. Kết thúc modul này, bạn sẽ có thể: (i) Mô tả các yêu cầu đối với việc tuân theo các quy định về chấp thuận sau khi thông hiểu; (ii) Mô tả tiến trình đạt được sự chấp thuận sau khi thông hiểu; (iii) Định nghĩa nhóm đối tượng dễ bị tổn thương; (iv) Mô tả các quy định về việc miễn trừ thủ tục chấp thuận sau khi thông hiểu. Các quy định về chấp thuận sau khi thông hiểu Các quy định tổng quátCó thể tìm đọc về các nguyên tắc căn bản cho sự chấp thuận sau khi thông hiểu trong Bộ Quy chế Liên bang, điều 45CFR 46.116(a) và 21CFR Phần 50.25(a). Một hồ sơ chấp thuận sau khi thông hiểu được coi là hợp pháp sẽ bao gồm các phần sau đây: 1.Một tuyên bố rằng cuộc thử nghiệm nhằm mục đích nghiên cứu, phần giải thích về các mục đích của cuộc nghiên cứu và khoảng thời gian thâm gia dự kiến cho đối tượng, phần mô tả về các thủ tục/thủ thuật phải thực hiện, và xác định rõ bất cứ thủ thuật nào còn trong vòng thí nghiệm. 2.Mô tả cho đối tượng biết về bất cứ rủi ro hoặc sự khó chịu nào có thể biết trước một cách hợp lý. 3.Mô tả cho đối tượng về bất cứ lợi ích nào mà đối tượng hoặc những người khác có khả năng hợp lý sẽ hưởng được từ cuộc nghiên cứu. 4.Thông báo về các thủ thuật hoặc quá trình điều trị thay thế thích hợp, nếu có, mà có thể có lợi cho đối tượng. 5.Một bản mô tả về phạm vi giữ bảo mật các hồ sơ có thể dùng để nhận diện đối tượng, nếu có, và lưu ý rằng có khả năng là Cơ Quan Quản Lý Thực Dược Phẩm có thể kiểm tra hồ sơ. 6.Đối với cuộc nghiên cứu có rủi ro nhiều hơn mức tối thiểu thì phải có phần giải thích về việc liệu có bất cứ khoản đền bù nào và có được điều trị y tế nào nếu xảy ra thương tích hay không và, nếu có, các khoản đó bao gồm những gì hoặc đối tượng có thể tìm thêm thông tin ở đâu. 7.Cung cấp thông tin cho đối tượng về người cần liên lạc để được giải đáp các câu hỏi có liên quan đến cuộc nghiên cứu và các quyền của đối tượng nghiên cứu, và người cần liên lạc trong trường hợp xảy ra thương tích có liên quan tới nghiên cứu. 8.Tuyên bố rằng việc tham gia là tự nguyện, việc từ chối tham gia sẽ không dẫn đến bị bất cứ hình phạt nào hoặc mất các quyền lợi mà đối tượng lẽ ra có quyền hưởng, và đối tượng có thể ngưng tham gia vào bất cứ lúc nào mà không bị phạt hoặc mất các quyền lợi mà lẽ ra đối tượng được hưởng. Các quy định khác Ngoài ra, các hồ sơ chấp thuận sau khi thông hiểu hợp pháp cũng cần bao gồm các yếu tố sau đây được nêu ra trong 45CFR 46.116(b) và 21CFR 50.25(b) Bộ Quy chế Liên bang nếu có liên quan đến nội dung nghiên cứu: 1.Tuyên bố rằng việc điều trị hoặc một thủ thuật cụ thể có thể có các rủi ro xảy ra cho đối tượng (hoặc cho phôi thai hoặc thai nhi, nếu đối tượng hiện đang hoặc có thể sẽ mang thai) mà hiện không tiên đoán trước được; 2.Các tình huống dự kiến xảy ra khiến nghiên cứu viên có thể quyết định chấm dứt sự tham gia của đối tượng mà không cần có sự chấp thuận của đối tượng; 3.Bất cứ chi phí thêm nào ma đối tượng có thể phải thanh toán khi tham gia cuộc nghiên cứu; 4.Các hậu quả của việc đối tượng quyết định rút ra khỏi cuộc nghiên cứu và các thủ tục dùng để chấm dứt sự tham gia của đối tượng một cách bài bản; 5.Tuyên bố rõ việc sẽ thông báo cho đối tượng những phát hiện mới đáng kể thu được từ quá trình nghiên cứu mà có thể ảnh hưởng đến sự sẵn lòng của đối tượng trong việc tiếp tục tham gia; 6.Số đối tượng ước tính tham gia trong cuộc nghiên cứu. Quy định về ClinicalTrials.Gov Cơ quan FDA cũng bổ sung thêm một yêu cầu mới có hiệu lực từ tháng 3 năm 2012 bên cạnh các yếu tố trước đây trong bản chấp thuận sau khi thông hiểu. Khi làm thủ tục chấp thuận sau khi thông hiểu cho các cuộc thử nghiệm lâm sàng đang thực hiện, như được định nghĩa trong 42 U.S.C.282(j)(1)(A), mỗi đối tượng tham gia thử nghiệm lâm sàng sẽ được thông báo thông tin sau đây trong các tài liệu và tiến trình chấp thuận sau khi thông hiểu. Thông báo này nhằm mục đích cho đối tượng thử nghiệm lâm sàng biết rằng thông tin thử nghiệm lâm sàng đã hoặc sẽ được chuyển nộp để cho vào cơ sở dữ liệu đăng ký về thử nghiệm lâm sàng theo đoạn (j) của phần 402 thuộc Đạo luật Dịch vụ sức khoẻ công cộng. "Sẽ có bài mô tả cuộc thử nghiệm lâm sàng này đăng trên http://www.ClinicalTrials.gov, theo yêu cầu của Luật pháp Mỹ. Trang mạng này sẽ không bao gồm thông tin có thể nhận diện bạn. Nhiều nhất thì trang mạng cũng sẽ chỉ đăng tải một bản tóm lược về kết quả. Bạn có thể truy tìm trang mạng này vào bất cứ lúc nào." Vào tháng 2 năm 2012, FDA cũng phát hành bản Hướng dẫn cho các nhà tài trợ, nghiên cứu viên và Ủy ban duyệt xét định chế - Các câu hỏi và trả lời về các yếu tố chấp thuận sau khi thông hiểu, 21CFR 50.25(c) Các nhóm Quản lý Việc tuân thủ các quy định về chấp thuận sau khi thông hiểu được quản lý bởi: ·FDA, 21 CFR 50 - xem phần Nghiên cứu theo Quy định của FDA ·DHHS, 45 CFR 46 - qua OHRP ·Các Ủy ban duyệt xét định chế (IRB) hiện hành. Nhận được sự chấp thuận sau khi thông hiểu Quá trình nhận được sự chấp thuận sau khi thông hiểu bao gồm: ·Cung cấp thông tin cho đối tượng. ·Trả lời các câu hỏi nào mà đối tượng có thể có để giúp họ hiểu vấn đề tốt hơn ·Nhận được sự chấp thuận tự nguyện của đối tượng để tham gia cuộc nghiên cứu. Cung cấp thông tin Hướng dẫn cung cấp thông tin bao gồm: ·Quảng cáo có thể là thông tin đầu tiên mà đối tượng được biết về nghiên cứu và thường được sử dụng nhiều nhất trong giai đoạn tuyển chọn. Các quảng cáo không được mang tính ép buộc hoặc đưa ra các hứa hẹn hoặc tuyên bố giả dối. Tiến trình chấp thuận sau khi thông hiểu thường được hiểu là bắt đầu ngay từ giai đoạn tuyển chọn. Ngoài ra, có một số lĩnh vực liên quan đến quá trình tuyển chọn nên xét tới như: oCác điều luật, hướng dẫn, hoặc chính sách của viện chi phối việc quảng cáo cho các đối tượng nghiên cứu, đặc biệt là loại nghiên cứu tại nhiều địa điểm; oViệc trả công cho những người tình nguyện tham gia nghiên cứu có được phép hay không tại nơi mà cuộc nghiên cứu được đề nghị, và điều đó sẽ được ghi nhận trong các tài liệu tuyển chọn đối tượng như thế nào; oCác "thông lệ" của quy trình tuyển chọn ở địa điểm cụ thể nơi diễn ra việc tuyển chọn và với nhóm đối tượng được đề nghị là gì. ·Các thủ tục để sàng lọc đối tượng tiềm năng xem họ có khả năng hội đủ điều kiện hay không phải bảo vệ các quyền và phúc lợi của đối tượng. ·Thông tin cần được truyền đạt theo một cách thức và bằng một ngôn ngữ rõ ràng và dễ hiểu, có hệ thống, và tạo điều kiện cho đối tượng đặt ra và được giải đáp các câu hỏi có thể có. ·Thông tin được truyền đạt không nên dùng ngôn từ có tính cách miễn giải trách nhiệm, cho dù trên văn bản chấp thuận hoặc trong các cuộc bàn luận về cuộc nghiên cứu. "Không một bản chấp thuận sau khi thông hiểu nào, cho dù bằng lời nói hay trên văn bản, được phép dùng bất cứ ngôn từ nào mang tính chất miễn giải trách nhiệm mà khiến cho đối tượng bắt buộc phải từ bỏ hoặc có vẻ từ bỏ bất cứ các quyền hợp pháp nào của đối tượng, hoặc miễn cho hoặc có vẻ như miễn cho nghiên cứu viên, nhà tài trợ, viện nghiên cứu, hoặc các đại lý của họ khỏi trách nhiệm về sự bất cẩn." 45CFR 46.116 Tăng cường kiến thứcNhững hướng dẫn về việc tăng cường kiến thức bao gồm: ·Cung cấp bản chấp thuận bằng ngôn ngữ dễ hiểu đối với đối tượng hoặc người đại diện của họ. ·Cung cấp cho các đối tượng không nói được tiếng Anh một bản chấp thuận sau khi thông hiểu đã được dịch chính xác theo quyết định của IRB. ·Nếu phải dùng một thông dịch viên thì vẫn cần phải cung cấp một bản dịch mẫu chấp thuận. Một số IRB cho phép sử dụng bản dịch tóm tắt tài liệu chấp thuận của IRB, xem 45CFR 46.117 (b)(2). ·Cho đối tượng có đủ thời gian để suy nghĩ về sự tham gia của mình trong cuộc nghiên cứu trước khi đưa ra sự chấp thuận. Nhận sự chấp thuận tham gia tự nguyệnBản chấp thuận sau khi thông hiểu có hiệu lực về pháp lý sẽ: ·Được lấy từ đối tượng hoặc người đại diện được úy quyền hợp pháp của đối tượng; ·Được lấy trong bối cảnh đã cho đối tượng có cơ hội cân nhắc kỹ việc có nên tham gia hay không và giảm thiểu các ảnh hưởng tạo áp lực. Không được sử dụng các chiến thuật tạo sự ép buôc như đền bù về tài chánh không thích hợp hoặc các phần thưởng khác; ·Không bao gồm bất cứ đoạn văn nào khiến đối tượng phải từ bỏ hoặc có vẻ như từ bỏ bất cứ các quyền hợp pháp nào của mình, hoặc miễn cho nghiên cứu viên, nhà tài trợ, hoặc viện nghiên cứu khỏi trách nhiệm pháp lý về sự bất cẩn; ·Các đối tượng nói tiếng Anh không biết đọc biết viết có thể "điểm chỉ" trên tài liệu chấp thuận sau khi thông hiểu, miễn là điều đó phù hợp với luật tiểu bang hiện hành. Các thách thức đặc biệt Vấn đề về ngôn ngữTiến trình chấp thuận sẽ được tiến hành bằng ngôn ngữ đối tượng sử dụng, và mẫu chấp thuận cần được dịch ra ngôn ngữ đó. Một IRB có thể yêu cầu sự xác nhận độc lập về tính chính xác của bản dịch. Các đối tượng nào không biết đọc biết viết bằng ngôn ngữ của họ phải có một thông dịch viên hiện diện để giải thích cuộc nghiên cứu cho đối tượng, và thông dịch các câu hỏi và trả lời giữa đối tượng và người lấy sự chấp thuận. Vấn đề về văn hoá Các vấn đề khác ngoài việc không biết đọc biết viết cũng có thể ảnh hưởng tới khả năng hiểu. Thí dụ, ở một số nền văn hoá, việc đặt câu hỏi đối với nhà nghiên cứu có thể được coi là mất lịch sự, hoặc việc từ chối những hành vi được cho là yêu cầu được giúp đỡ cũng là không lịch thiệp. Trong các trường hợp này, vấn đề ai là người tiến hành quy trình lấy sự chấp thuận và cách giải thích quy trình này như thế nào thậm chí còn quan trọng hơn. Nhóm người dễ bị tổn thương Khái niệm về tính dễ tổn thương của đối tượng rất quan trọng đối với đạo đức nghiên cứu và việc tuân thủ theo quy định. Quy định 21CFR 56.111(b) của FDA yêu cầu rằng "khi một số hoặc tất cả đối tượng rất có thể dễ bị tổn thương do sự ép buộc hoặc bị ảnh hưởng không chính đáng, thì phải có các biện pháp bảo vệ được bổ sung vào cuộc nghiên cứu để bảo vệ cho các quyền và phúc lợi của những đối tượng này". Các quy định này không định nghĩa thuật ngữ "đối tượng dễ bị tổn thương" hoặc giải thích về các nguyên nhân có thể gây ra sự tổn thương, nhưng có cung cấp một danh sách các thí dụ sau đây về những đối tượng dễ bị tổn thương: "trẻ em, tù nhân, phụ nữ mang thai, người tật nguyền, người bị khiếm khuyết về tâm thần hoặc người bị thiệt thòi về kinh tế hoặc giáo dục." Các thí dụ khác bao gồm: ·Bệnh nhân trong các tình huống khẩn cấp; ·Các đối tượng ở bên lề xã hội (thí dụ, người đồng tình nam/đồng tình nữ/người lưỡng tính/người chuyển giới, những công nhân không có giấy tờ); ·Các thành viên của một nhóm có cấu trúc thứ bậc, như sinh viên y khoa, dược khoa, nha khoa, nhân viên điều dưỡng cấp dưới tại một bệnh viện hay phòng thí nghiệm, và các thành viên trong lực lượng vũ trang; ·Các bệnh nhân bị căn bệnh nguy hiểm chết người hoặc không thể chữa khỏi; ·Những người già; ·Người trong viện điều dưỡng; ·Những người bị thất nghiệp hoặc nghèo khó; ·Các nhóm thiểu số sắc tộc; ·Những người vô gia cư, người du mục, người tị nạn. Mặc dù cụm từ "dễ bị tổn thương" không được định nghĩa rõ trong các quy định, nhưng coi đây là một giới hạn về quyền tự quyết mang đến một công cụ quý báu để suy xét xem đã có đủ các biện pháp bảo vệ hay chưa trong một dự án nghiên cứu nhằm bảo vệ quyền và phúc lợi cho các đối tượng này. Các Quy định miễn trừ sự chấp thuận sau khi thông hiểuĐôi khi, trong các trường hợp cụ thể, việc chấp thuận sau khi thông hiểu có thể được miễn nếu có sự chấp thuận của IRB. Các Quy định của DHHS về việc miễn trừ DHHS (45 CFR 46.116) cho phép IRB miễn trừ hoặc thay đổi các yêu cầu đối với việc chấp thuận sau khi thông hiểu trong hai trường hợp sau: 1.Các dự án của Chính phủ; 2.Các miễn trừ và thay đổi tổng quát. Các Dự án của Chính phủNếu nghiên cứu/dự án được chấp thuận và/hoặc được tiến hành bởi chính phủ tiểu bang hay địa phương và nhằm mục đích nghiên cứu/đánh giá: ·Một chương trình đem lại ích lợi cho công chúng. ·Các thủ tục trong các chương trình này. ·Các lựa chọn khác ngoài các chương trình hoặc thủ tục này. ·Các thay đổi về phương pháp thanh toán hoặc mức độ dịch vụ của các chương trình này. ·Nhìn về thực tế, cuộc nghiên cứu sẽ không thể được tiến hành nếu không được miễn trừ. Yêu cầu được miễn trừ và thay đổi tổng quát có thể được chấp nhận nếu IRB xác định:·Cuộc nghiên cứu không có mức rủi ro cao hơn mức rủi ro tối thiểu cho đối tượng. ·Sự miễn trừ hoặc thay đổi sẽ không có ảnh hưởng bất lợi đối với các quyền và phúc lợi của đối tượng. ·Cuộc nghiên cứu sẽ không thể được tiến hành nếu không được miễn hoặc thay đổi. ·Mỗi khi thích hợp, các đối tượng sẽ được cung cấp thêm thông tin có liên quan sau khi tham gia. Các Quy định của FDA về Những trường hợp được miễn thực hiện yêu cầu về chấp thuận sau khi thông hiểu FDA 21 CFR 50.23 và 21 CFR 50.24 cho phép miễn thực hiện các yêu cầu đối với việc chấp thuận sau khi thông hiểu trong các trường hợp sau: ·Trong các tình huống đáp ứng các yêu cầu được miễn việc lấy sự chấp thuận sau khi thông hiểu đối với trường hợp nghiên cứu khẩn cấp (21CFR 50.24). ·Trong các tình huống mà tính mạng của một đối tượng đang bị đe dọa, các yêu cầu để được miễn lấy sự chấp thuận sau khi thông hiểu được đáp ứng đầy đủ, và nghiên cứu viên có hồ sơ chứng minh cho tất cả những điều sau đây: oSau khi hội ý với một bác sĩ khác, nghiên cứu viên tin rằng tình huống dẫn đến việc phải dùng một sản phẩm thử nghiệm (thí dụ, một loại thuốc, dụng cụ, hoặc sản phẩm sinh học còn trong vòng thí nghiệm); oĐối tượng và/hoặc đại diện được ủy quyền hợp pháp không có khả năng biểu lộ sự chấp thuận (Định nghĩa của FDA về đại diện được Ủy quyền hợp pháp: một cá nhân, một tổ chức tư pháp hay một đoàn thể khác được ủy quyền theo luật pháp hiện hành để chấp thuận thay mặt cho một đối tượng tiềm năng về việc tham gia của đối tượng đó trong (các) thủ tục/thủ thuật có liên quan tới nghiên cứu); oKhông có đủ thì giờ để lấy sự chấp thuận. oKhông có lựa chọn nào khác ngang bằng hoặc tốt hơn để cứu mạng sống của đối tượng. Các trường hợp được miễn ký mẫu chấp thuận/ Chấp thuận bằng lời nói Có thể được miễn lấy văn bản chấp thuận có chữ ký và nhận sự chấp thuận bằng lời nói trong các trường hợp sau đây: FDA MỹCơ quan FDA của Mỹ cho phép miễn lập hồ sơ chấp thuận sau khi thông hiểu (21CFR 56.109(c)[1]) khi việc tham gia nghiên cứu có mức rủi ro tối thiểu đối với đối tượng và không có liên quan gì tới các thủ tục đòi hỏi sự chấp thuận bên ngoài phạm vi của việc tham gia cuộc nghiên cứu. IRB có thể yêu cầu nghiên cứu viên cung cấp cho đối tượng các văn bản tài liệu về cuộc nghiên cứu. DHHSTheo DHHS (45CFR 46.117), IRB có thể cho phép miễn lập văn bản chấp thuận có chữ ký ở một trong các tình huống sau đây: ·Bản chấp thuận sẽ là nối kết duy nhất giữa cuộc nghiên cứu và đối tượng và rủi ro chính đối với đối tượng là có thể bị xâm phạm thông tin bảo mật, và mỗi đối tượng sẽ được hỏi liệu họ có muốn lập hồ sơ chấp thuận hay không. ·Việc tham gia cuộc nghiên cứu có mức rủi ro tổi thiểu về nguy hại đối với đối tượng và cuộc nghiên cứu không có liên quan gì tới các thủ tục đòi hỏi sự chấp thuận bên ngoài phạm vi của việc tham gia cuộc nghiên cứu. ·Nghiên cứu viên có thể được IRB yêu cầu cung cấp một bản tóm lược về cuộc nghiên cứu cho đối tượng nếu một trong những phương pháp này được sử dụng. Việc lấy chấp thuận qua điện thoại từ 1 đại diện được ủy quyền hợp pháp thích hợp? FDA Sự chấp thuận bằng lời nói không thỏa mãn quy định trong 21CFR 56.109(c) là phải có văn bản chấp thuận có chữ ký phù hợp với các yêu cầu trong 21CFR 50.27(a). Tuy nhiên, có thể chấp nhận được việc gửi tài liệu chấp thuận sau khi thông hiểu cho đại diện được ủy quyền hợp pháp (Legally Authorized Representative_LAR) qua fax và tiến hành cuộc phỏng vấn lấy lời chấp thuận qua điện thoại để LAR có thể vừa đọc bản chấp thuận vừa bàn bạc về nó. Nếu LAR đồng ý, người này có thể ký vào bản chấp thuận và gửi lại văn bản có chữ ký cho nghiên cứu viên lâm sàng qua fax.
|